P6
Disease-tailored therapeutic strategies against hyperinflammatory viral infections caused by highly pathogenic respiratory viruses


Summary
Zoonotic transmission of highly pathogenic respiratory viruses such as corona- and influenza viruses into a naïve human population poses a serious pandemic threat, which was impressively demonstrated by the recent SARS-CoV-2 pandemic. Human infections with such highly pathogenic respiratory viruses often result in life threatening systemic inflammatory diseases that include extensive pulmonary damage and respiratory failure, vascular dysfunction and multiple organ damage as signs of viral sepsis. The responsible virus-induced immune responses are characterized by delayed or perturbed induction of the early antiviral innate immune response, which culminates in imbalanced and unproportional high expression levels of inflammatory mediators in the later stages of the disease. To unravel these virus-specific immune response patterns as well as the virus-specific triggers, and provide effective antiviral as well as anti-inflammatory treatment options in the future, our project aims to dissect the virus- and cell type-specific dynamics of the early immune responses and lead to the inflammatory disease outcome by using in vivo models of related highly and low pathogenic viruses.
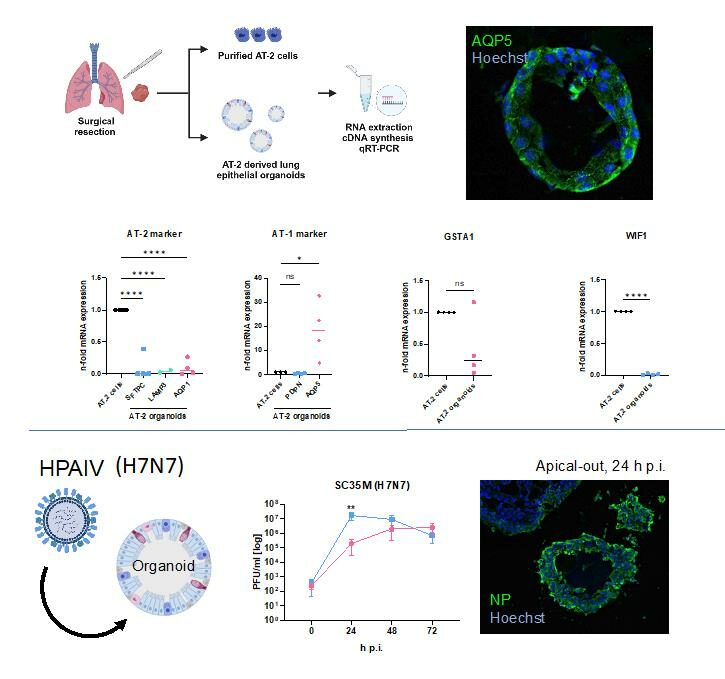
Furthermore, this project builds up on the results of the first funding period and aims to dissect the crucial involvement of the MAPK p38 signaling pathway for the immune responses during COVID-19 and infections with highlight pathogenic influenza viruses as well as to unravel its promising potential as a target for anti-inflammatory as well as advanced antiviral treatment strategies using two pre-clinically tested p38 inhibitors. Employing advanced primary tissue models and alveolar organoids of the human lung, this project is strongly focused to convert the gained knowledge into new and improved clinical treatment strategies for infections with highly pathogenic respiratory viruses now and in the future.


