P7
Multidimensional profiling of novel mediators and therapeutic targets related to AKI during systemic inflammation


Summary
Acute kidney injury (AKI) is a common complication in critically ill patients with sepsis. AKI has a significant impact on the short- and long-term course of the patient’s disease and is associated with a significantly higher mortality rate. As there is no sufficient therapeutic option (except for renal replacement therapy in patients with severe AKI as a purely supportive measure), prevention is of great importance. Remote ischemic preconditioning (RIPC) consists of defined episodes of ischemia and reperfusion before the final damage to the target organ occurs. We have already shown that the application of RIPC (3 cycles of 5 minutes each) before cardiac surgery significantly reduces the occurrence of AKI in high-risk patients (37.5% versus 52.5% in the control group; p = 0.02). In addition, we observed that an early and transient increase (before surgery) in the two G1 cell cycle arrest markers TIMP2 and IGFBP7 was associated with a reduced incidence of AKI. Furthermore, patients showed significantly increased HMGB1 (High Mobility Group Box 1) concentrations in their urine early on after the application of RIPC compared to the control group. HMGB1 is an alarmin from the group of DAMP molecules (Damage Associated Molecular Pattern), which is released in response to cell stress. These molecules act as signal mediators to trigger endogenous reactions, e.g. activation of the innate immune system. A preliminary experimental study has already shown that HMGB1 application prior to an injury event can attenuate AKI. It was also shown that this effect led to an upregulation of Sema5b through the activation of AMPKα and NF-κB, which in turn induces a transient, protective cell cycle arrest.
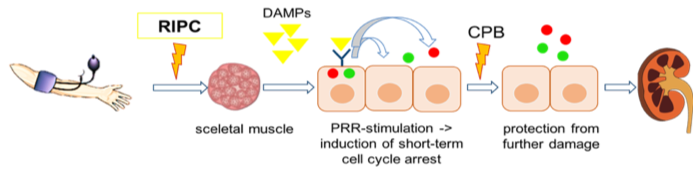
Conceptual model for RIPC. The application of RIPC releases damage associated molecular patterns (DAMPs) from the skeletal muscle cells. These DAMPs get filtered in the glomerulus and subsequently bind to the pattern recognition receptors (PRRs) on renal tubular epithelial cells. Consequently, a short-term cell cycle arrest is induced which protects the cells from further damage (e.g. ischemia reperfusion injury through CPB).
The aim of the current project is now to transfer the results we have obtained from the cardiac surgery cohort to critically ill patients with early sepsis. In addition, we would like to gain further insights into the pathophysiological background by using renal organoid models. The following three objectives are pursued: Aim 1: Validation and mechanistic analysis of novel potential mediators and therapeutic targets in AKI and RIPC. Aim 2: Molecular characterization of the role of Bst2 in the pathogenesis of AKI. Aim 3: Investigate the role of RIPC on SA-AKI


