P1
Targeting nociceptors to modulate neutrophil mobilization and homing in sepsis


Summary
Sepsis is a life-threatening immune reaction triggered by the invasion of (mostly bacterial) pathogens into the blood stream. Patients suffering from sepsis exhibit hypotension, tissue edema and impaired microcirculation leading to tissue hypoxia, organ dysfunction and high mortality. At the cellular level, sepsis induces endothelial dysfunction resulting in a loss of endothelial barrier integrity. Although preclinical investigations suggest promoting endothelial function during sepsis can lower sepsis complications, the underlying mechanisms are incompletely understood and no therapeutic tool is available to limit endothelial dysfunction and improve microcirculatory flow during sepsis.
Procalcitonin is a 116 amino acid hormokine ubiquitously expressed by immune and parenchymal cells during sepsis. Procalcitonin’s plasma concentrations increase up to 100.000-fold during sepsis and correlate with sepsis severity and outcome. Hence, procalcitonin is a clinically well characterized biomarker for the diagnosis, treatment and prognosis of patients with sepsis but it is yet unknown whether and how procalcitonin itself functions in sepsis. Our previous studies and preliminary data show that procalcitonin itself is a mediator of endothelial dysfunction. Importantly, procalcitonin induces loss of endothelial barrier integrity by reducing endothelial surface levels of vascular endothelial-cadherin (VE-cadherin) resulting in tissue edema in vivo. Full length procalcitonin (PCT1-116) is cleaved by dipeptidyl peptidase IV (DPPIV) to PCT3-116 and our preliminary data show that cleavage of PCT1-116 by DPPIV triggers the barrier-disrupting activity.
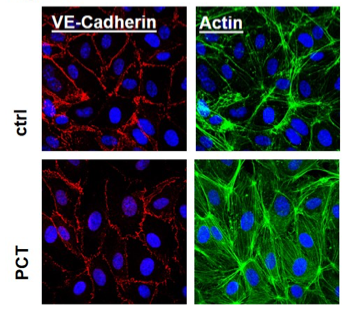
Procalcitonin (PCT) increases endothelial permeability and induces VE-cadherin phosphorylation and internalization. Immunohistochemistry of 1ng/ml PCT or vehicle-treated HUVECs (Human Umbilical Vein Endothelial Cells) shows that PCT induces re-distribution of VE-cadherin and actin stress fiber Formation.
We thus hypothesize that PCT3-116 is a DPPIV-generated trigger of sepsis-induced vascular barrier dysfunction and that DPPIV can serve as a therapeutic target to preserve vascular integrity during systemic inflammation by limiting the presence of active procalcitonin. We will test this hypothesis by identifying the putative procalcitonin receptor on endothelial cells, characterizing the procalcitonin-induced signaling pathways and analyzing the role of VE-cadherin as a downstream target of procalcitonin.
We will also investigate the effects of DPPIV inhibitors on procalcitonin dynamics and hemodynamics of the micro- and macrocirculation in septic pigs and we will conduct a pilot study on the use of DPPIV inhibitors to reduce procalcitonin levels, limit microcirculatory dysfunction and modulate the incidence of a systemic inflammatory response syndrome in patients undergoing on-pump cardiac surgery. Our goal is to characterize the role of procalcitonin as a potent inductor of endothelial barrier dysfunction and to establish clinically licensed DPPIV inhibitors as a potential therapeutical option to preserve endothelial integrity, microcirculatory flow and organ perfusion during systemic inflammation associated with hyperprocalcitonemia.


